
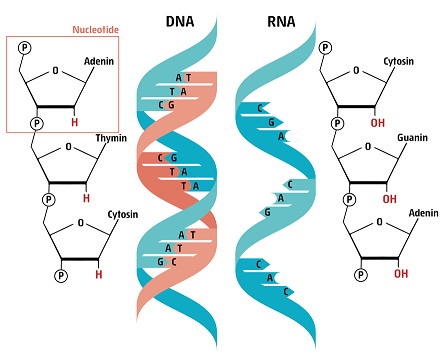
The building blocks of the human genome are four different types of nucleotides linked one after the other in a long chain. Two such chains (blue and orange on our figure) join together to make up the shape of a giant twisted ladder of the DNA. Every nucleotide contains one of four different bases: Adenine (A), Guanine (G), Cytosine (C) or Thymine (T), that make up the “rungs” or “steps of this giant twisted “ladder”.
Similar to a book, where the letters encode words and sentences, the information of the DNA is hidden in the sequence of the nucleotides. When our body “reads” the letters, different proteins will be made from different sequences.
Similarly, when “typos” are made in the genetic material, which we call mutations, it is possible that the characteristics of the encoded proteins will change. (Learn more about DNA structure and how its code is translated) SARS-CoV-2, the virus causing COVID-19 also uses nucleotides to store genetic information, but its letters are written in a different molecular ladder: RNA. Although very similar to DNA, RNA has only one chain and is chemically less stable than DNA.
When we sequence the viral genome, we want to “read” the whole RNA sequence containing 30,000 nucleotide letters.
In a first step the RNA has to be isolated from the human sample. Next, the RNA is copied into DNA. This means that we generate DNA, that has the same sequence of letters as the original RNA molecule. Human cells cannot make DNA from RNA, so for this, we need to use special enzymes, that can only be found in some viruses.
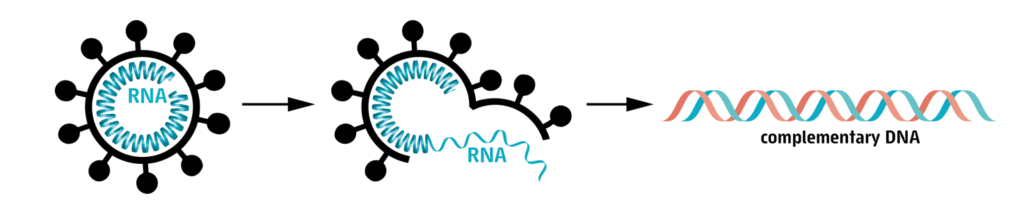
Then, the DNA is amplified by a method called polymerase chain reaction, or PCR (Learn more about PCR).
A common diagnostic PCR only amplifies one small ~120 nucleotide-long piece of the viral genome to detect the presence of virus. As we want to know the sequence of the entire genome, we have to amplify each and every part of the 30,000-letters-long sequence. At the end of this step, we have thousands and thousands of small DNA fragments that are identical copies of different parts of the viral RNA genome.
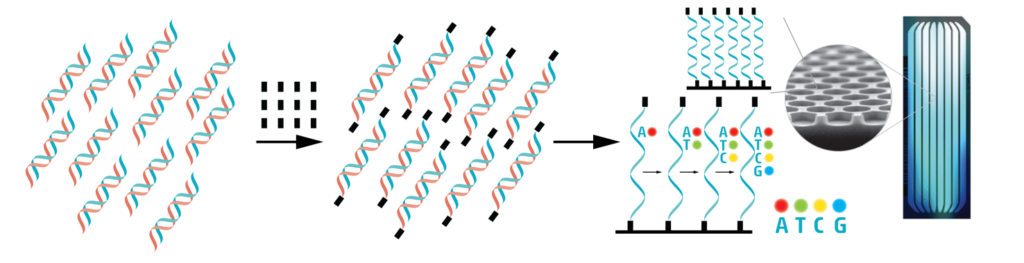
Adaptor sequences represented by black squares are added to the end of these small fragments. Then the fragments are loaded on a small glass chip. Here, they attach with the help of the adaptor sequences. On these chips, the attached fragments will be copied again, but the nucleotide letters that are used to make the new copies are labelled with fluorescent molecules. Each letter fluoresces in a different color, and the colors are used to read which letter was added. As the copies are built by adding nucleotides, images are taken at each step, and the colors tell us one by one what nucleotide was added (Learn more about sequencing).
The so-called “Next-generation sequencing” is capable of elucidating the sequence of millions of nucleotides in a short time. Today, a human genome, that is more than 3 billion nucleotides long, only takes a couple of days to sequence, while the same task took 13 years when it was performed the first time, from 1990 to 2003!
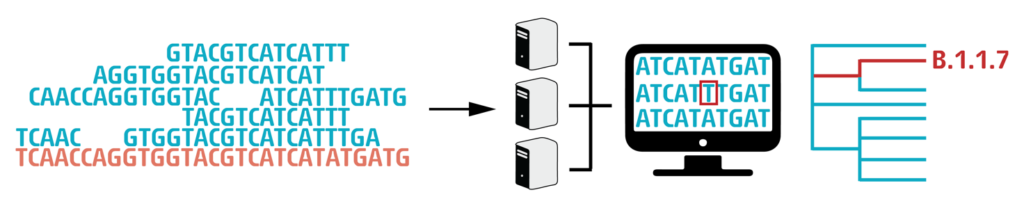
The technique used by CeMM for Next-generation sequencing can only read DNA fragments of 200-500 nucleotides at a time. These short “reads” then have to be stitched together like a puzzle to get the entire, 30,000-long sequence. This is done by using a “reference genome” (orange on our figure), the first ever sequenced SARS-CoV-2 genome, that was isolated in December 2019 in Wuhan, China. When the whole 30.000 nucleotide long “puzzle” is put together, we can finally compare it to the reference genome and see where differences (mutations) can be found.
Finally, we analyze the sequences by bioinformatic tools. With this information, the spread of the virus and the accumulation of mutations can be monitored. This allows for better tracing of transmission chains as well as understanding changes in the properties of the virus.
Each time DNA or RNA is copied, there is a chance that an error (“Mutation”) will occur. For DNA replications, the specific enzyme that copy DNA, called polymerase, can proofread and fix errors, which results in fewer mutations. RNA polymerase on the other hand lacks this skill for the most part, leading to a higher mutation rate in RNA viruses.
Coronaviruses have a lower mutation rate than many other RNA viruses, as they are capable of some light error-correction. Most of these mutations are harmful for the virus itself and vanish quickly, however some of them give the virus an advantage and will be carried into the next virus generation. After a while the mutation becomes a “fixed” part of the genome in a virus population. The amount of fixed mutation over time is called the fixation rate and gives a sense of how fast the virus is evolving.
For SARS-CoV-2 it is estimated that about every 11 days a new fixed mutation emerges. See Martin et al., Science 371, Vol. 6528 (2021)
This is a coding mutation in the genome of SARS-CoV-2 which alters the spike protein. D614G emerged in January 2020 and since then is found in many virus strains worldwide. The majority of the Austrian SARS-CoV-2 genomes sequenced by us from end of February/March onwards contained this mutation already.
Laboratory experiments showed that the mutation stabilises the spike protein, which is present as a trimer on the surface of the virion, and improves the binding to the cellular receptor ACE2. These studies suggest that the mutation may confer increased infectivity to SARS-CoV-2 in the human population.
RNA viruses are prone to random mutations in their genomes and accumulate continuously new mutations as drivers of their evolution. In our study by Popa, Genger et al., we investigated cases from a transmission chain with 8 transmission events and were able to observe the establishment of a new mutation. We observed that this mutation appeared first as a minority variant with 3.6% in one individual.
After transmission, this virus mutant reached a frequency of 25% in one case and established as a “fixed” (100%) mutation in two other infects. The particular observed non-coding mutation has no effect on the protein sequence of the virus but we cannot predict its impact on the overall virus fitness.
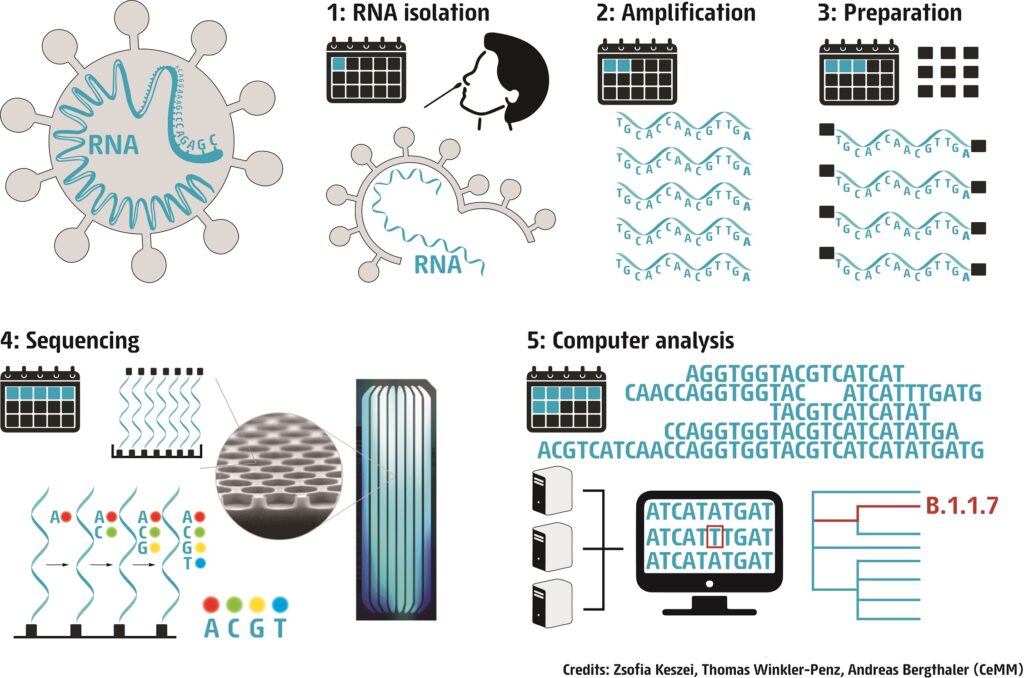
Here is an illustration to demonstrate that a standard next-generation sequencing and data analysis pipeline for SARS-CoV-2 viral genomes takes approximately seven days (Credits: Zsofia Keszei, Thomas Winkler-Penz, Andreas Bergthaler / CeMM). Yes, there are other methods that are faster, but our aim is to obtain viral full-length genomes with low frequency variants at high quality. See also our publication Popa et al. Science Translational Medicine 2020.