
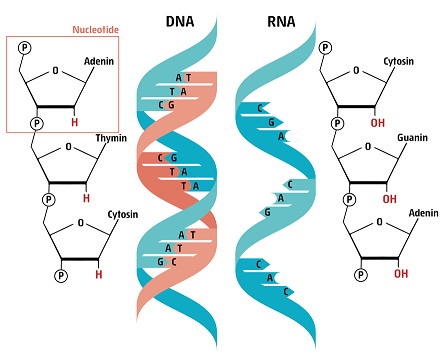
Die Bausteine des menschlichen Genoms bestehen aus vier verschieden Arten von Nukleotiden, die wie an einer Kette einer nach dem anderen miteinander verbunden sind. Zwei solcher Ketten (blau und orange in unserer Grafik) bilden zusammen die gewundene Doppelhelix-struktur unserer DNS. Jeder einzelne Nucleotid beinhalten einer von vier unterschiedlichen Basen: Adenin (A), Guanin (G), Cytosin (C) oder Thymin (T), diese bilden die „Sprossen“ der gewundenen DNS-„Leiter“.
Ähnlich wie in einem Buch, in dem Buchstaben Wörter und Sätze bilden, die wir lesen und verstehen können, ist die genetische Information der DNS in der Abfolge der Nukleinbasen kodiert. Das Auslesen unterschiedlicher Basenabfolgen führt zu der Bildung von unterschiedlichen Proteinen basierend auf den unterschiedlichen Sequenzen. Entstehen während der Vermehrung der DNS Fehler in dieser Reihenfolge, kann es zu einer strukturellen und funktionalen Änderung des kodierten Proteins führen. Man nennt solche „Tippfehler“ innerhalb des Erbguts „Mutationen“.
Hier finden Sie weiterführende Video über DNS-Struktur und wie die Information ausgelesen und übersetzt wird.
SARS CoV-2, das Coronavirus der die Krankheit COVID-19 verursacht, kodiert seine Erbinformation ebenfalls als Abfolge von Nucleotiden, allerdings in einer anderen Form der molekularen „Leiter“: Ribonukleinsäure (RNS). Obwohl ähnlich wie DNS, besteht RNS nur aus einem Strang und ist chemisch instabiler als DNS.
Bei einer Ganzgenomsequenzierung wird die gesamte RNS Sequenz des Virus ausgelesen, diese besteht im Falle von SARS-CoV-2 aus ungefähr 30,000 einzelne Nukleotide.
Im ersten Schritt muss die RNS des Virus vom menschlichen Material der Probe getrennt oder isoliert werden. Anschließend wird die RNS in DNS umgeschrieben, also eine DNS-Sequenz künstlich generiert, welche dieselbe Abfolge von Nukleinbasen aufweist, wie die originale RNS-Sequenz. Da menschliche Zellen nicht in der Lage sind RNS auf DNS umzuschreiben, werden für diesen Prozess spezielle Enzyme benötigt, die nur in einigen Viren zu finden sind.
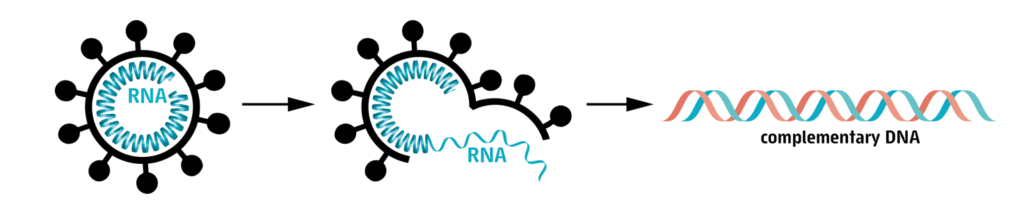
Im nächsten Schritt wird die generierte DNS mittels der Polymerase-Kettenreaktion (PCR) vervielfältig. Hier ein kurzes Erklärungsvideo.
Ein PCR Lauf für diagnostische Zwecke vervielfältigt normalerweise nur ein kleines ~120 Nukleotid langes Stück eines viralen Genoms, um die Präsenz des Virus in einer Probe detektieren zu können. Um Änderungen innerhalb des gesamten Genoms untersuchen zu können und nicht nur innerhalb eines kleinen Bereiches, müssen alle 30,000 Nukleotide vervielfältigt werden. Am Ende dieses Prozesses sind mehrere tausend kleiner DNS-Fragmente entstanden, welche identische Kopien verschiedener Bereiche des viralen RNS Genoms sind.
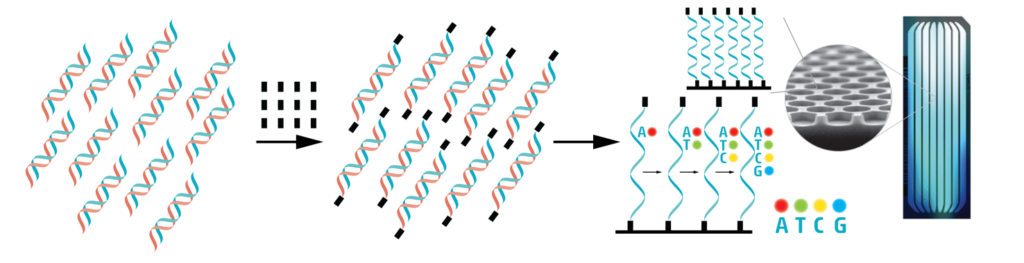
Adaptersequenzen (in unserer Grafik als schwarze Rechtecke dargestellt) werden hinzugefügt und verbinden sich mit den Enden der DNS-Fragmente. Diese werden dann auf einen kleinen Glas Chip transferiert, wo sie sich mit Hilfe der Adaptersequenzen anheften. Auf diesem Chip werden alle so fixierte Fragmente noch einmal vervielfältigt.
Die einzelnen Nukleinbasen, welche in die Kopien eingebaut werden, sind mit fluoreszierenden Molekülen verbunden, welche, je nach Base, in einer anderen Farbe leuchten. Nach jedem Schritt im Kopiervorgang wird ein Bild angefertigt und die Farbe zeigt an, welche Base an dieser Position hinzugefügt wurde.
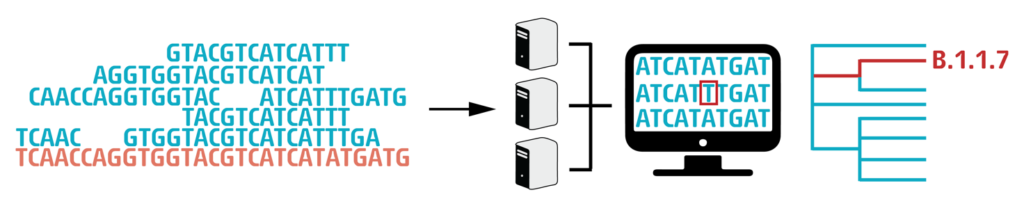
Die „Next-Generation“ Sequenzierungsmethode, die von CeMM eingesetzt wird, kann lediglich 200-500 Nukleotid lange DNS-Fragmente auslesen. Diese kurzen „reads“, müssen anschließend wieder in der richtigen Reihenfolge zusammengefügt werden, um die gesamte 30,000 Nukleotid lange Virusgenomsequenz zu erhalten. Hierzu werden die einzelnen Stücke mit einem Referenzgenom verglichen (hier in unserer Grafik in Orange). Hierfür dient das erste jemals sequenzierte Genom von SARS-CoV-2, isoliert im Dezember 2019 in Wuhan, China. Nachdem alle Fragmente den richtigen Positionen am Genom zugeordnet wurden, kann die neue Sequenz mit der Referenzsequenz verglichen werden und Unterschiede (Mutationen) in den Sequenzen können gefunden werden.
Nun wird die Sequenz mittels bioinformatischer Analysen untersucht, um weitere Erkenntnisse (zum Beispiel die phylogenetische Verwandtschaft zwischen verschiedenen Sequenzen) gewinnen zu können. Mit diesen Informationen kann die Ausbreitung des Virus und das Auftreten neuer Mutationen überwacht werden. Dies erlaubt sowohl eine verbesserte Nachverfolgung von Infektionsketten als auch ein besseres Verständnis von Veränderungen des Virus.
Jedes Mal, wenn DNS oder RNS kopiert wird, gibt es die Chance, dass während des Kopiervorganges ein Fehler („Mutation“) passiert. DNS-Replikation ist weniger fehleranfällig als RNS Replikation, da die Enzyme, welche DNS kopieren die Fähigkeit besitzen Fehler zu erkennen und zu korrigieren. Dies fehlt bei der RNS Replikation, was zu höheren Fehlerraten führt als bei DNS.
Die meisten dieser Mutationen schaden dem Virus und verschwinden daher schnell wieder. Ab und zu kann es aber passieren, dass eine Mutation zufällig eine positive Wirkung für das Virus hat und an die nächste Generation von Viren weitergegeben wird. Nach und nach werden immer mehr Viren diese Mutation tragen und der „Fehler“ ist nun ein fixer Bestanteil des Virengenoms. In diesem Fall spricht man von einer „fixierten“ Mutation. Die Geschwindigkeit, in der neue fixierte Mutationen entstehen, wurde bei SARS CoV-2 auf etwa eine neue, fixierte Mutation etwa alle 11 Tage geschätzt. See Martin et al., Science 371, Vol. 6528 (2021)
Dies ist für einen RNS-Virus eine eher niedrige Rate, denn Coronaviren, zu denen auch SARS-CoV-2 zählt, besitzen im Vergleich zu anderen RNS – Viren Mechanismen, welche eine leicht korrigierende Funktion haben und daher eine geringe Anfälligkeit neue Mutationen zu bilden.
Zur oft gestellten Frage, ob das SARS-CoV-2 Virus schon mutiert ist, haben wir ein 12-minütiges Video vorbereitet.
Es handelt sich hier um ein System, um Mutationen innerhalb der S-region von SARS-CoV-2 zu beschreiben. Mutationen in diesem Bereich verändern die Sequenz, welche für das Spike-Protein codiert, welches als herausragende Proteinstruktur an der Virushülle an den menschlichen Zellrezeptor ACE2 bindet und so eine wichtige Rolle bei der Infizierung der Wirtszelle spielt.
D614G entstand in Jänner 2021 und findet sich in vielen verschieden Viruslinien weltweit. Die Mehrzahl aller österreichischen Virengenome, die von CeMM seit Februar/ März 2020 sequenziert wurden, trugen diese Mutation bereits. In Experimenten unter Laborbedingungen zeigte diese Mutation eine stabilisierende Wirkung auf das Spike-Protein und erleichterte die Bindung an den ACE2 Rezeptor. Diese Studien legen nahe, dass diese Mutation zu einer erhöhten Infektiosität von SARS-CoV-2 beim Menschen beiträgt.
RNS Viren sind anfällig für zufällige Mutationen innerhalb ihres Genoms und häufen so kontinuierlich neue Mutationen an als Triebkraft ihrer Evolution. In unserer Studie by Popa, Genger et al. konnten Infektionsketten mit bis zu 8 individuellen Infektionsereignissen nachverfolgt werden. In diesen Fällen war es uns möglich das Aufkommen und die Fixierung einer neuen Mutation zu beobachten. Wir zeigten, dass die Mutation zum ersten Mal nur in einer Minderheit (3,6%) aller Virusgenomen innerhalb eines Infektionsfalles auftauchte.
Nach der Infektion einer weiteren Person, fand sich diese Mutation nun in bis zu 25% aller Virusgenome dieser Person und konnte sich „fixieren“ (100% aller Virusgenome trugen diese Mutation) in zwei späteren Infektionsfällen. Diese spezielle Mutation führt nicht zu einer Änderung eines Proteins, die Auswirkungen auf die „Gesamtfitness“ des Virus kann aber nicht vorhergesagt werden.
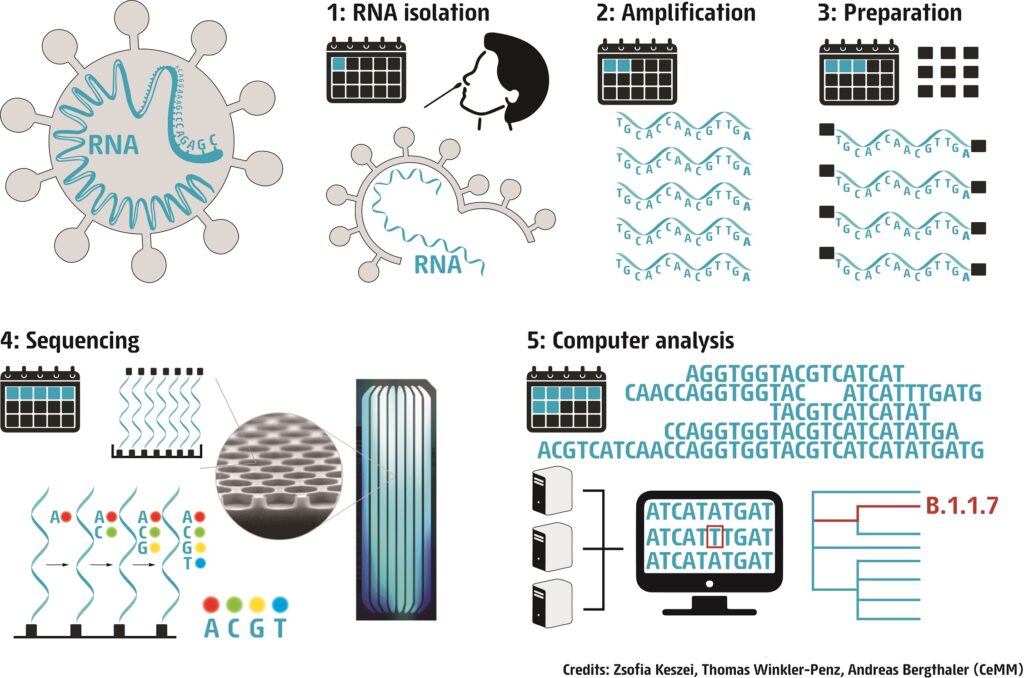
Folgende Grafik zeigt die einzelnen Schritte, um eine Ganzgenomsequenzierung durchführen zu können. Von RNS Isolation bis hin zu den finalen Endergebnissen dauert der gesamte Prozess im Durchschnitt sieben Tage. Credits: Zsofia Keszei, Thomas Winkler-Penz, Andreas Bergthaler / CeMM). Es gibt andere Methoden, die zu schnelleren Ergebnissen führen.
Unser Ziel sind allerdings hoch-qualitative Sequenzen des gesamten Virengenoms auch von niedrig-frequenten Varianten. Weitere Informationen finden Sie in unserer Studie Popa et al. Science Translational Medicine 2020.